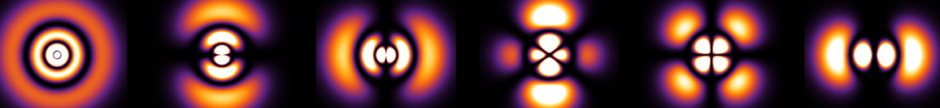
The purpose of this post is to explain why, experimentally, one only observes \(4\) electric dipole transitions in the IR spectrum of buckminsterfullerene, also known as \(\text C_{60}\) or informally as the buckyball:

Buckyball Basics
The simplest conceptual way to construct a buckyball is to start with a regular icosahedron:
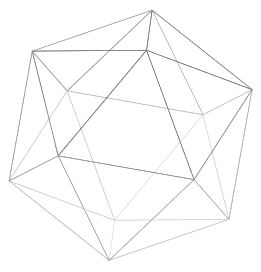
which has \(F=20\) equilateral triangular faces, \(V=12\) vertices, and \(E=30\) edges; notice this obeys Euler’s formula \(F+V=E+2\). Then, by simply “shaving off” each of the \(V=12\) vertices; it is clear from the picture these \(12\) vertices would transform into \(12\) pentagonal faces, each surrounded by \(5\) hexagonal faces so that no \(2\) pentagonal faces share an adjacent edge, yielding a buckyball topology (and geometrically, all \(\text C-\text C\) covalent bonds should be of equal length). This shaving process increases the number of faces to \(F’=32\) (of which \(12\) are pentagonal and \(20\) are hexagonal), the number of vertices to \(V’=60\) (i.e. just the number of carbon atoms), and the number of edges to \(E’=90\) such that Euler’s formula is maintained \(F’+V’=E’+2\).
IR Spectroscopy Selection Rules
Recall that within the Born-Oppenheimer approximation, the \(N\) nuclei of some molecule are “clamped” at positions \(\textbf X:=(\textbf X_1,…,\textbf X_N)\) and are regarded as moving in the effective potential \(V_{\text{eff}}(\textbf X)\) due to the electrons \(e^-\), so that the molecular Hamiltonian is:
\[H=T_{\text n}+V_{\text{eff}}(\textbf X)\]
If in addition one approximates \(V_{\text{eff}}(\textbf X)\) by a harmonic potential about the (stable) equilibrium configuration \(\textbf X_0\) of the nuclei:
\[V_{\text{eff}}(\textbf X)\approx\frac{1}{2}(\textbf X-\textbf X_0)^T\left(\frac{\partial^2 V_{\text{eff}}}{\partial\textbf X^2}\right)_{\textbf X_0}(\textbf X-\textbf X_0)\]
then, upon diagonalizing the Hessian \(\left(\frac{\partial^2 V_{\text{eff}}}{\partial\textbf X^2}\right)_{\textbf X_0}\) into the orthonormal eigenbasis of its normal modes, one obtains \(3N\) decoupled simple harmonic oscillators so the spectrum of \(H\) is just that of an anisotropic harmonic oscillator in \(\textbf R^{3N}\), at least within all the assumptions made so far (e.g. ignoring anharmonicity of \(V_{\text{eff}}(\textbf X)\)). Each Fock eigenstate \(|n_1,n_2,…,n_{3N}\rangle\) of \(H\) (thought of as a vibrational eigenstate because one can view \(H\) as a Hamiltonian governing vibrations/SHM of the nuclei about equilibrium) is thus a product \(|n_1,n_2,…,n_{3N}\rangle=\otimes_{i=1}^{3N}|n_i\rangle\) of suitable \(1\)D quantum harmonic oscillator wavefunctions.
IR radiation is just like any other electromagnetic radiation in that (within the dipole approximation, which is fair for long-wavelength IR radiation despite the larger size of molecules) it stimulates electric dipole transitions via the time-dependent perturbation \(\Delta H=\Delta H(t)\) to the molecular Hamiltonian \(H\):
\[\Delta H=-\boldsymbol{\pi}\cdot\textbf E_0\cos \omega_{\text{IR}} t\]
where the electric dipole moment \(\boldsymbol{\pi}=\boldsymbol{\pi}(\textbf X,\textbf x)\) of the molecule is:
\[\boldsymbol{\pi}:=e\sum_{\text{nuclei }i}Z_i\textbf X_i-e\sum_{\text{electrons }i}\textbf x_i\]
and the molecule is assumed to be neutral \(\sum_{\text{nuclei }i}Z_i=\sum_{\text{electrons }i}\). By Fermi’s golden rule, the molecular transition rate between two distinct vibrational \(H\)-eigenstates \(|n_1,…,n_{3N}\rangle\to|n’_1,…,n’_{3N}\rangle\) is proportional to the mod-square of the matrix element of the (time-independent amplitude of the) perturbation:
\[|\langle n’_1,…,n’_{3N}|\boldsymbol{\pi}\cdot\textbf E_0|n\rangle|^2\]
However, most IR spectroscopy experiments (e.g. IR laser sources in FTIR spectrometers) use unpolarized IR radiation, so this means one should really replace by an isotropic averaging factor of \(1/3\) and forget about (as far as selection rules are concerned) a factor of \(|\textbf E_0|^2\), so just focus on:
\[|\langle n’_1,…,n’_{3N}|\boldsymbol{\pi}|n\rangle|^2\]
Similar to what was done earlier for the effective potential \(V_{\text{eff}}(\textbf X)\), one can also Taylor expand the dipole moment operator \(\boldsymbol{\pi}\) within the configuration space \(\textbf X\) of the nuclei about the equilibrium configuration \(\textbf X_0\):
\[\boldsymbol{\pi}\approx\boldsymbol{\pi}(\textbf X_0)+\left(\frac{\partial\boldsymbol{\pi}}{\partial\textbf X}\right)_{\textbf X_0}(\textbf X-\textbf X_0)+…\]
Sandwiching this in the matrix element, the constant term \(\boldsymbol{\pi}(\textbf X_0)\) (which represents the possibility of a permanent electric dipole like in a water molecule) vanishes by orthogonality of the distinct vibrational \(H\)-eigenstates, so the quantity of interest is just:
\[\left|\left(\frac{\partial\boldsymbol{\pi}}{\partial\textbf X}\right)_{\textbf X_0}\langle n’_1,…,n’_{3N}|\textbf X-\textbf X_0|n_1,…,n_{3N}\rangle\right|^2\]
This immediately leads to a “gross selection rule” for two vibrational \(H\)-eigenstates to be coupled by the IR perturbation \(\Delta H\), namely that the Jacobian \(\left(\frac{\partial\boldsymbol{\pi}}{\partial\textbf X}\right)_{\textbf X_0}\) must be non-vanishing; physically, this means that only the normal modes of the nuclear vibration that experience a change in \(\boldsymbol{\pi}\) when the nuclei are slightly displaced from \(\textbf X_0\) will be IR-active.
…main point of this section is to explain that IR transitions are low-energy excitations require a change in electric dipole moment b/w initial and final states, which, factoring out the charge e, means the matrix element of the position observable b/w two states must be non-zero by Fermi’s golden rule. There are actually \(2\) selection rules for IR spectroscopy; the gross selection rule is that \(\partial\boldsymbol{\pi}/\partial\) must be zero.
Character Tables of Finite Group Representations
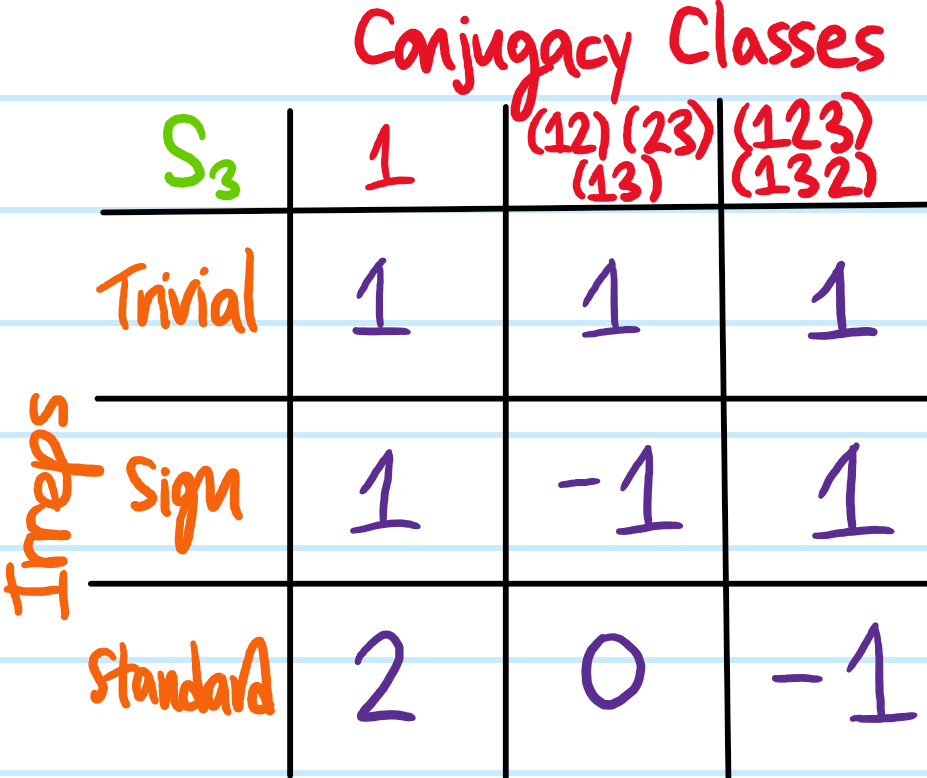
As a warmup, consider the symmetric group \(S_3=\{1, (12), (13), (23), (123), (132)\}\) of order \(|S_3|=3!=6\). Then \(S_3\) can be partitioned into \(3\) conjugacy classes, namely \(S_3=\{1\}\cup\{(12), (13), (23)\}\cup\{(123), (132)\}\). Each \(S_3\) conjugacy class maps onto an \(S_3\)-irrep, so there will also be three \(S_3\)-irreps. One of them is always just the trivial irrep whereby the permutations do nothing to all vectors. For symmetric groups, there is also always the sign irrep \(1,(123),(132)\mapsto 1\) and \((12),(13),(23)\mapsto -1\) such that odd permutations do nothing to all vectors but even \(A_3\)-permutations flip vectors across the origin. Finally, there is the standard \(S_3\)-irrep that one might intuitively think about as acting on the vertices of an equilateral triangle which rigidly drags the whole Cartesian plane \(\textbf R^2\) along with it. Note the dimensions of these \(S_3\)-irreps are the only ones that could have been compatible with its order \(6=1^2+1^2+2^2\).
Each \(S_3\)-irrep is associated with its own character class function which can be evaluated on an arbitrary representative of each conjugacy class. For instance, the trivial irrep is \(1\)-dimensional and its character always evaluates to \(1\) on all conjugacy classes. The sign irrep is also \(1\)-dimensional but its character evaluated on each conjugacy class is instead the sign of the permutations in that conjugacy class. Finally, noting that \(\cos(2\pi/3)=-1/2\) and that the trace of a rotation in \(\textbf R^2\) by angle \(\theta\) is \(2\cos\theta\), the following character table for \(S_3\) may be obtained:
So far this example has been fairly abstract. To bridge this “abstract nonsense” with chemistry, consider the specific example of the ammonia molecule \(\text{NH}_3\):

In this case, rather than pulling a random group (like \(S_3\)) out of thin air, here one can use the ammonia molecule to motivate studying the specific group of actions on \(\textbf R^3\) that leave it looking like nothing happened. In chemist jargon, one is interested in the point group of the ammonia molecule, the adjective “point” implying that any such action on \(\textbf R^3\) must have at least one fixed point in space (commonly chosen to be the origin) so that e.g. translations of the ammonia molecule are disregarded (cf. the distinction in special relativity between the Poincare group and its Lorentz subgroup). Note that this is indeed a group thanks to its very definition (if one action leaves ammonia looking like nothing happened, and a second action also leaves ammonia looking like nothing happened, then composing them will also leave ammonia looking like nothing happened).
So what is the point group of the ammonia molecule? For this, there isn’t really any super rigorous/systematic way to do it, one just has to stare hard at the molecule and think…
Clearly, one way to leave ammonia looking like nothing happened is to literally do nothing. There is also manifestly \(C_3\) rotational symmetry (by \(120^{\circ}\) or \(240^{\circ}\)). Finally, there are \(3\) reflection symmetries about “vertical” mirror planes. As a mathematician, it is thus easy to recognize that the point group of the ammonia molecule is just the dihedral group \(D_3\) (which happens to be isomorphic to \(S_3\)). However, as a chemist one would instead refer to this as the “\(C_{3v}\) point group”, where the \(C\) and the \(3\) subscript are meant to emphasize the \(C_3\) subgroup of rotational symmetries mentioned above, while the “\(v\)” subscript is meant to emphasize that the mirror planes are “vertical”. Strictly speaking, one should also check that there are no horizontal mirror planes, inversion centers, or improper rotation axes in order to be able to confidently assert that the point group of ammonia really is \(C_{3v}\) rather than \(C_{3v}\) merely being a subgroup of an actually larger point group.
The isomorphism \(C_{3v}\cong D_3\cong S_3\) means that by happy chance the character table for the ammonia point group \(C_{3v}\) is already known. For instance, the transpositions \((12),(23),(13)\) in \(S_3\) map onto vertical mirror plane reflections in \(C_{3v}\), while the \(3\)-cycles \((123),(132)\) are simply \(C_3\) rotations. Indeed, the \(3\)-dimensional \(C_{3v}\)-representation on the ammonia molecule itself embedded rigidly in \(\textbf R^3\) is reducible into a direct sum of the trivial irrep acting on the \(1\)-dimensional \(z\)-axis and the standard irrep acting on the \(2\)-dimensional \(xy\)-plane. It is also worth verifying that the columns of the character table satisfy orthonormality \(\sum_{\text{ccl}\text{ of }C_{3v}}|\text{ccl}|\chi_{\phi}(\text{ccl})\chi_{\phi’}(\text{ccl})=|C_{3v}|\delta_{\phi\cong\phi’}\) for any \(2\) \(C_{3v}\)-irreps \(\phi,\phi’\).
One can also consider how various scalar-valued and vector-valued polynomials on \(\textbf R^3\) transform passively under the \(C_{3v}\) point group. For instance, any function \(f=f(\rho)\) of the cylindrical coordinate \(\rho:=\sqrt{x^2+y^2}\) only, such as \(f(\rho)=\rho^2\), will transform under the trivial \(C_{3v}\)-irrep.
Making the reasonable assumption that all \(60\) carbon atoms are specifically of the \(^{12}\text C\) isotope, and are therefore identical bosons, so something about the permutation group \(S_{60}\)? (formalize the earlier comment about “looking the same” via the idea of identical quantum particles/ bosons and fermions where in that case the vectors are not in \(\textbf R^3\) but state vectors in the symmetrized tensor product Hilbert space of identical bosons)